- Serce Dziecka
- Wszystko o sercu
- Wady serca
- Zespół niedorozwoju lewego serca
Zespół niedorozwoju lewego serca
HLHS
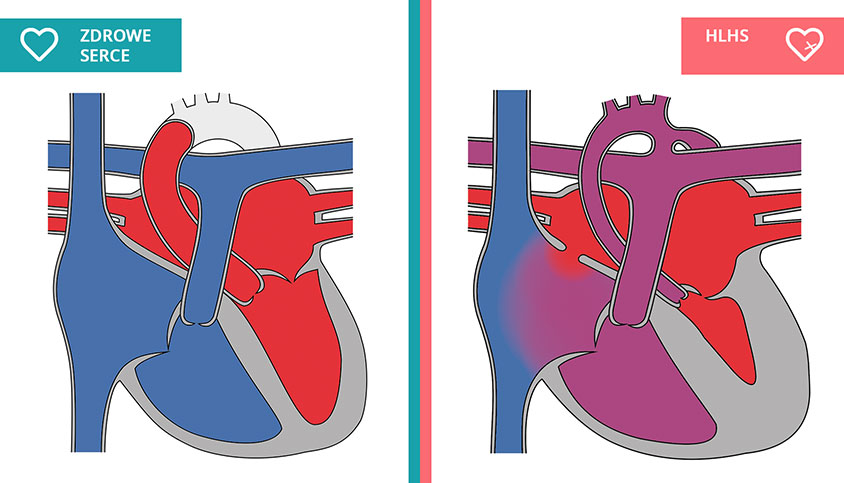
Na czym polega wada
Pojęcie „zespół niedorozwoju lewego serca” (hypoplastic left heart syndrome – HLHS) obejmuje zwężenie lub zarośnięcie zastawki dwudzielnej/mitralnej, różnego stopnia niedorozwój lub brak lewej komory, zwężenie lub zarośnięcie zastawki aortalnej oraz niedorozwój aorty wstępującej i łuku aorty. Wada ta stanowi 1,4–8,6% wrodzonych wad serca.
Zespół ten jest najczęstszą postacią serca jednokomorowego. W 20–80% przypadków towarzyszy mu zwężenie cieśni aorty.
Przyczyny powstawania wady nie zostały dotychczas wyjaśnione. Wydaje się, że najbardziej prawdopodobna jest teoria dotycząca pierwotnej nieprawidłowości zastawki aortalnej – jej zwężenia lub zarośnięcia. Brak przepływu przez zastawkę aorty w okresie życia płodowego nie stymuluje rozwoju lewej komory, zastawki dwudzielnej, a także aorty wstępującej i łuku aorty. Teorię tę potwierdza obserwacja prawidłowego rozwoju lewej komory w przypadku współistnienia dużego ubytku w przegrodzie międzykomorowej.
W okresie życia płodowego dziecko z HLHS rozwija się normalnie. Noworodki z tą wadą są najczęściej donoszonymi dziećmi o prawidłowej urodzeniowej masie ciała. W przeciwieństwie do innych wad serca tej wadzie rzadko towarzyszą anomalie rozwojowe innych narządów.
Noworodek z zespołem niedorozwoju lewego serca ma funkcjonalnie jedną komorę zaopatrującą krążenie płucne i krążenie systemowe. Przeżycie dziecka po urodzeniu jest uzależnione od drożności przewodu tętniczego oraz obecności otworu owalnego. Rozpoczęcie wymiany gazowej w płucach i uruchomienie krążenia płucnego wpływa na znaczny spadek oporu w krążeniu płucnym i zwiększenie przepływu płucnego kosztem systemowego. Wzrost oporu systemowego (zamykanie się przewodu tętniczego oraz odcięcie łożyska), a przede wszystkim dalszy spadek oporu płucnego w pierwszych dniach życia przyczyniają się do stopniowego zmniejszania się przepływu systemowego. Konsekwencją przeciążenia objętościowego i ciśnieniowego prawej komory jest szybko postępująca niewydolność krążenia. Około 95% nieleczonych dzieci umiera w 1. miesiącu życia. Przed erą leczenia chirurgicznego HLHS prowadził do 25–30% zgonów u dzieci w pierwszym tygodniu życia z przyczyn związanych z układem krążenia i nadal jest najczęstszą przyczyną zgonów noworodków z wadami serca.
Objawy
Objawy zespołu niedorozwoju lewego serca pojawiają się zwykle 24–48 godzin po urodzeniu. Przewaga przepływu płucnego nad systemowym przyczynia się do rozwoju zespołu małego rzutu serca (tachykardia, zimne kończyny, upośledzenie powrotu włośniczkowego, oliguria i anuria, kwasica metaboliczna). Pojawia się duszność stanowiąca próbę wyrównania kwasicy metabolicznej. Dalsze zwiększenie przepływu płucnego prowadzi do rozwoju stanu zagrożenia życia dziecka. Tętno na tętnicach udowych jest zwykle bardzo słabo wyczuwalne. Sinica występuje rzadko.
Leczenie
Postępowanie przedoperacyjne prowadzi się w celu utrzymania równowagi przepływów w krążeniu płucnym i systemowym dzięki utrzymaniu drożności przewodu tętniczego oraz sterowania oporem naczyniowym płuc i krążenia obwodowego. Podawanie prostaglandyny E1 (PGE1) zapobiega zamykaniu się przewodu tętniczego i zapewnia przepływ systemowy, nie likwidując jednak objętościowego obciążenia komory.
Opór w krążeniu systemowym obniża się, stosując leki rozszerzające naczynia (np. nitroprusydek sodu). Często praca serca wymaga wsparcia i zastosowania leków inotropowych (dopamina). Pomimo tych wysiłków okres przedoperacyjny cechuje duża niestabilność stanu dziecka.
Jedyny sposób leczenia dzieci z zespołem niedorozwoju lewego serca to leczenie operacyjne, przeszczepienie serca lub wieloetapowa korekcja.
Rozpoznanie zespołu niedorozwoju lewego serca stanowi bezwzględne wskazanie do leczenia operacyjnego i wobec śmiertelnego przebiegu naturalnego tej wady nie istnieją przeciwwskazania. W ośrodkach, w których wykonuje się przeszczepianie serca, przeciwwskazaniem do wieloetapowego leczenia rekonstrukcyjnego jest znacznego stopnia niedomykalność zastawki pnia płucnego lub zastawki trójdzielnej, słaba funkcja skurczowa prawej komory czy też małe rozmiary aorty ( Końcowy etap rekonstrukcyjnego leczenia złożonych wad serca o typie pojedynczej komory stanowi operacja Fontana. Podstawowym warunkiem jej powodzenia są dobrze rozwinięte naczynia płucne i mały opór w krążeniu płucnym. Znaczny opór w krążeniu płucnym u noworodka uniemożliwia zastosowanie tego rozwiązania w pierwszych miesiącach życia. Operacją przygotowującą układ krążenia dziecka z zespołem niedorozwoju lewego serca do operacji Fontana jest operacja Norwooda.
Obecnie u dzieci z zespołem niedorozwoju lewego serca powszechnie stosuje się 3-etapowa korekcję. W wieku noworodkowym wykonuje się operację Norwooda, w 4.–6. miesiącu życia operację hemi-Fontana, a pomiędzy 18. i 24. miesiącem zmodyfikowaną operację Fontana (opisy – patrz serce jednokomorowe).

Celem operacji Norwooda jest umożliwienie normalnego dojrzewania krążenia płucnego, zapewnienie niezależnego od przewodu tętniczego przepływu systemowego oraz zmniejszenie obciążenia pojedynczej komory. Operację przeprowadza się w krążeniu pozaustrojowym i hipotermii głębokiej z zatrzymaniem krążenia.
Pierwszy etap operacji to wycięcie fragmentu przegrody międzyprzedsionkowej w celu wytworzenia swobodnej komunikacji międzyprzedsionkowej. Następnie likwiduje się przewód tętniczy i poszerza aortę, wszywając łatę z homogennej tętnicy płucnej. Zrekonstruowaną aortę zespala się z pniem płucnym, tworząc w ten sposób wypływ z pojedynczej komory do krążenia systemowego. Przepływ krwi w krążeniu płucnym zapewnia zespolenie (rurka z tworzywa sztucznego) pomiędzy przednią ścianą prawej komory a tętnicą płucną.
Zmodyfikowaną operację Norwooda wykonuje się od końca lat 90. ubiegłego stulecia (w ośrodku krakowskim od 2001 roku). Podstawową różnicę stanowi inna lokalizacja bliższego odcinka zespolenia systemowo-płucnego. W operacji zmodyfikowanej zespolenie wychodzi z pojedynczej komory, a nie, jak w operacji klasycznej, stanowi odgałęzienie pnia ramienno-głowowego lub tętnicy podobojczykowej.

Zaletą nowego typu zespolenia jest mniejszy wpływ na ciśnienie rozkurczowe w aorcie i w konsekwencji lepszy i bardziej stabilny przepływ w krążeniu wieńcowym. U dzieci po operacji zmodyfikowanej stwierdza się bardziej stabilny wczesny przebieg pooperacyjny.
Powikłania
Śmiertelność po pierwszym etapie leczenia operacyjnego, zwłaszcza w okresie wczesnym, jest nadal bardzo duża (20–45%) i decydująco wpływa na ostateczne wyniki wieloetapowego leczenia. Do najczęstszych późnych powikłań należą nawrót zwężenia cieśni aorty oraz zwężenie lewej tętnicy płucnej. Zwężenie cieśni aorty koryguje się, wykonując balonową plastykę w czasie cewnikowania serca przed kolejnym etapem leczenia operacyjnego. Zwężenie lewej tętnicy płucnej usuwa się w czasie operacji hemi-Fontana.
